- 1. Getting Started
-
2.
First Semester Topics
-
General Chemistry Review
- Introduction
- Electron Configurations of Atoms
- QM Description of Orbitals
- Practice Time - Electron Configurations
- Hybridization
- Closer Look at Hybridization
- Strategy to Determine Hybridization
- Practice Time! - Hybridization
- Formal Charge
- Practice Time - Formal Charge
- Acids-bases
- Practice Time - Acids and Bases
- Hydrogen Bonding is a Verb!
- Progress Pulse
-
Structure and Bonding
- Chemical Intuition
- From Quantum Mechanics to the Blackboard: The Power of Approximations
- Atomic Orbitals
- Electron Configurations of Atoms
- Electron Configurations Tutorial
- Practice Time - Structure and Bonding 1
- Lewis Structures
- Drawing Lewis Structures
- Valence Bond Theory
- Valence Bond Theory Tutorial
- Hybridization
- Polar Covalent Bonds
- Formal Charge
- Practice Time - Structure and Bonding 2
- Curved Arrow Notation
- Resonance
- Electrons behave like waves
- MO Theory Intro
- Structural Representations
- Progress Pulse
-
Acids/Base and Reactions
- Reactions
- Reaction Arrows: What do they mean?
- Thermodynamics of Reactions
- Acids Intro
- Practice Time! Generating a conjugate base.
- Lewis Acids and Bases
- pKa Scale
- Practice Time! pKa's
- Predicting Acid-Base Reactions from pKa
- Structure and Acidity
- Structure and Acidity II
- Practice Time! Structure and Acidity
- Curved Arrows and Reactions
- Nucleophiles
- Electrophiles
- Practice Time! Identifying Nucleophiles and Electrophiles
- Mechanisms and Arrow Pushing
- Practice Time! Mechanisms and Reactions
- Energy Diagrams and Reactions
- Practice Time! - Energy Diagrams
- Progress Pulse
- Introduction to Retrosynthesis
-
Alkanes and Cycloalkanes
- Introduction to Hydrocarbons and Alkanes
- Occurrence
- Functional Groups
- Practice Time! Functional Groups.
- Structure of Alkanes - Structure of Methane
- Structure of Alkanes - Structure of Ethane
- Naming Alkanes
- Practice Time! Naming Alkanes
- Alkane Isomers
- Relative Stability of Acyclic Alkanes
- Physical Properties of Alkanes
- Ranking Boiling Point and Solubility of Compounds
- Conformations of Acyclic Alkanes
- Practice Time! Conformations of acyclic alkanes.
- Conformations of Cyclic Alkanes
- Naming Bicyclic Compounds
- Stability of Cycloalkane (Combustion Analysis)
- Degree of Unsaturation
-
Stereochemistry
- Enalapril in ACE
- Constitutional and Stereoisomers
- Chirality or Handedness
- Drawing a Molecules Mirror Image
- Exploring Mirror Image Structures
- Enantiomers
- Drawing Enantiomers
- Practice Time! Drawing Enantiomers
- Identifying Chiral Centers
- Practice Time! Identifying Chiral Molecules
- CIP (Cahn-Ingold-Prelog) Priorities
- Determining R/S Configuration
- Diastereomers
- Meso Compounds
- Fischer Projections
- Fischer Projections: Carbohydrates
- Measuring Chiral Purity
- Practice Time! - Determining Chiral Purity and ee
- Chirality and Drugs
- Chiral Synthesis
- Prochirality
- Converting Fischer Projections to Zig-zag Structures
- Practice Time! - Assigning R/S Configurations
-
Alkenes and Addition Reactions
- The Structure of Alkenes
- Alkene Structure - Ethene
- Physical Properties of Alkenes
- Naming Alkenes
- Health Insight - BVO (Brominated Vegetable Oil)
- E/Z and CIP
- Stability of Alkenes
- H-X Addition to Alkenes: Hydrohalogenation
- Practice Time - Hydrohalogenation
- X2 Addition to Alkenes: Halogenation
- HOX addition: Halohydrins
- Practice Time - Halogenation
- Hydroboration/Oxidation of Alkenes: Hydration
- Practice Time - Hydroboration-Oxidation
- Oxymercuration-Reduction: Hydration
- Practice Time - Oxymercuration/Reduction
- Oxidation and Reduction in Organic Chemistry
- Calculating Oxidation States of Carbon
- Identifying oxidation and reduction reactions
- Practice Time - Oxidation and Reduction in Organic
- Oxidation
- Reduction
- Capsaicin
-
Alkynes
- Structure of Ethyne (Acetylene)
- Naming Alkynes
- Practice Time! - Naming Alkynes
- Physical Properties of Alkynes
- Preparation of Alkynes
- Practice Time! - Preparation of Alkynes
- H-X Addition to Alkynes
- X2 Addition
- Hydration
- Reduction of Alkynes
- Practice Time! - Addition Reactions of Alkynes
- Oxidative Cleavage of Alkynes
- Alkyne Acidity and Acetylide Anions
- Reactions of Acetylide Anions
- Retrosynthesis Revisted
- Practice Time! - Multistep Synthesis Using Acetylides
-
Alkyl Halides and Alcohol
- Naming Alkyl Halides
- Naming Alcohols
- Classes of Alcohols and Alkyl Halides
- Practice Time! - Naming Alkyl Halides
- Practice Time! - Naming Alcohols
- Physical Properties of Alcohols and Alkyl Halides
- Structure and Reactivity of Alcohols
- Structure and Reactivity of Alkyl Halides
- Preparation of Alkyl Halides and Tosylates from Alcohols
- Practice Time! - Alcohols to Alkyl Halides
- Preparation of Alkyl Halides from Alkenes; Allylic Bromination
- Strategy for Predicting Products of Allylic Brominations
- Practice Time! - Allylic Bromination
-
Substitutions (SN1/SN2) and Eliminations (E1/E2)
- Introduction
- Solvents
- SN1 Reaction: The Carbocation Pathway
- SN2 Reactions: The Concerted Backside Attack
- SN1 vs. SN2: Choosing the Right Path
- Application: Cardura (Doxazosin)
- E1 Reactions: Elimination via Carbocations
- E2 Reactions: The Concerted Elimination
- E1cB: The Conjugate Base Elimination Pathway
- Substitution versus Elimination
- Dienes, Allylic and Benzylic systems
-
General Chemistry Review
-
3.
Second Semester Topics
- Arenes and Aromaticity
-
Reactions of Arenes
- Electrophilic Aromatic Substitution
- EAS-Halogenation
- EAS-Nitration
- Practice Time - Synthesis of Aniline
- EAS-Alkylation
- Practice Time - Friedel Crafts Alkylation
- EAS-Acylation
- Practice Time - Synthesis of Alkyl Arenes
- EAS-Sulfonation
- Practice Time - EAS
- Donation and Withdrawal of Electrons
- Regiochemistry in EAS
- Practice Time - Directing Group Effects
- Synthesizing Disubstituted Benzenes: Effects of Substituents on Rate and Orientation
- Steric Considerations
- Strategies for Synthesizing Disubstituted Benzenes
- NAS - Addition/Elimination
- NAS - Elimination/Addition - Benzyne
- Alcohols and Phenols
-
Ethers and Epoxides
- Intro and Occurrence
- Crown Ethers and Cryptands
- Preparation of Ethers
- Reactions of Ethers
- Practice Time - Ethers
- Preparation of Epoxides
- Reactions of Epoxides - Acidic Ring Opening
- Practice Time - Acidic Ring Opening
- Reactions of Epoxides - Nucleophilic Ring Opening
- Practice Time - Nucleophilic Ring opening
- Application - Epoxidation in Reboxetine Synthesis
- Application - Nucleophilic Epoxide Ring Opening in Crixivan Synthesis
- Physical Properties of Ethers and Epoxides
- Naming Ethers and Epoxides
-
Aldehydes and Ketones
- Naming Aldehydes and Ketones
- Physical Properties of Ketones and Aldehydes
- Practice Time - Naming Aldehydes/Ketones
- Nucleophilic addition
- Addition of Water - Gem Diols
- Practice Time - Hydration of Ketones and Aldehydes
- Addition of Alcohols - Hemiacetals and Acetals
- Acetal Protecting Groups
- Hemiacetals in Carbohydrates
- Practice Time - Hemiacetals and Acetals
- Addition of Amines - Imines
- Addition of Amines - Enamines
- Practice Time - Imines and Enamines
- Application - Imatinab Enamine Synthesis
- Addition of CN - Cyanohydrins
- Practice Time - Cyanohydrins
- Application - Isentress Synthesis
- Addition of Ylides - Wittig Reaction
- Practice Time - Wittig Olefination
- Structure of Ketones and Aldehydes
- Carboxylic Acids and Derivatives
- Enols and Enolates
- Condensation Reactions
-
4.
NMR, IR, UV and MS
- Spectroscopy
-
HNMR
- Nuclear Spin
- Interpreting
- Chemical Shift
- Practice Time! - Chemical Shift
- Equivalency
- Indentifying Homotopic, Enantiotopic and Diastereotopic Protons
- Practice Time! - Equivalency
- Intensity of Signals
- Spin Spin Splitting
- Practice Time! - Spin-Spin Splitting
- Primer on ¹³C NMR Spectroscopy
- Alkanes
- Alkynes
- Alcohols
- Alkenes
- Coupling in Cis/Trans Alkenes
- Ketones
- HNMR Practice 1
- HNMR Practice 2
- HNMR Practice 3
- HNMR Practice 4
- Exchangeable Protons and Deuterium Exchange
- IR - Infrared Spectroscopy
- UV - Ultraviolet Spectroscopy
- Mass Spectrometry
-
5.
General Chemistry
- General Chemistry Lab
- Strategy for Balancing Chemical Reactions
- Calculator Tips for Chemistry
- Significant Figures
- Practice Time! Significant Figures
- Spreadsheets - Getting Started
- Spreadsheets - Charts and Trend lines
- Standard Deviation
- Standard Deviation Calculations
- Factor Labels
- Practice Time! - Factor Labels
- Limiting Reagent Problem
- Percent Composition
- Molar Mass Calculation
- Average Atomic Mass
- Empirical Formula
- Practice time! Empirical and Molecular Formulae
- Initial Rate Analysis
- Practice Time! Initial Rate Analysis
- Solving Equilibrium Problems with ICE
- Practice Time! Equilibrium ICE Tables
- Le Chatelier's
- Practice Time! Le Chatelier's Principle
- 6. Organic Chemistry Lab
- 7. Tools and Reference
-
8.
Tutorials
- Reaction Mechanisms (introduction)
- Factor Labels
- Acetylides and Synthesis
- Drawing Cyclohexane Chair Structures
- Drawing Lewis Structures
- Aromaticity Tutorial
- Common Named Aromatics (Crossword Puzzle)
- Functional Groups (Flashcards)
- Characteristic Reactions of Functional Groups
- Alkyl and Alkenyl Groups
- Valence Bond Theory
- Alkane Nomenclature
-
9.
The Alchemy of Drug Development
- Ivermectin: From Merck Innovation to Global Health Impact
- The Fen-Phen Fix: A Weight Loss Dream Turned Heartbreak
- The Asymmetry of Harm: Thalidomide and the Power of Molecular Shape
- Semaglutide (Ozempic): From Lizard Spit to a Once-Weekly Wonder
- From Cocaine to Novocain: The Development of Safer Local Anesthesia
- The Crixivan Saga: A Targeted Strike Against HIV
- The story of Merck’s COX-2 inhibitor, Vioxx (rofecoxib)
- The Accidental Aphrodisiac: The Story of Viagra
- THC: A Double-Edged Sword with Potential Neuroprotective Properties?
- Ritonavir Near Disaster and Polymorphism
- 10. Allied Health Chem
- 11. Sandbox
Clear History
Ligand Substitution
Introduction
Ligand substitution reactions involve the replacement of coordinated ligands in transition metal complexes as shown by figure 1. These reactions are fundamental in both stoichiometric and catalytic processes, often serving as the rate-determining step. They are usually an essential first step.
MLx+ nL' ⇌ MLx-n L’n + nL
Figure 1: General ligand substitution reaction scheme: L is the leaving ligand and L' is the incoming ligand.
Mechanisms of Ligand Substitution
Substitution mechanisms in organometallic chemistry resemble organic SN1 and SN2 reactions, following two primary pathways:
-
Associative (A or SN2 like)
-
The incoming ligand binds first, forming a higher-coordinate intermediate.
-
Common in 16-electron complexes (e.g., square planar d8 complexes of Ni(II), Pd(II), Pt(II)).
- Classic associative ligand substitution reaction with second order kinetics, characteric of 16-electron transition metal complexes (Figure 2).
-
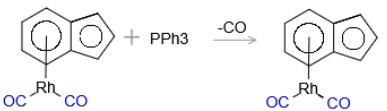
Figure 2: Second-order kinetics in the associative substitution of (η⁵-carbocycle)Rh(CO)₂ with PPh₃, following the rate law: d[product]/dt = k₂[(η⁵-carbocycle)Rh(CO)₂][PPh₃]
-
Dissociative (D or SN1 like)
-
A ligand dissociates first, creating a lower-coordinate intermediate before the new ligand binds.
-
Typical for 18-electron complexes, which must lose a ligand to become reactive.
- Classic example of dissociative substitution in 18-electron, tetrahedral d¹⁰ complexes (Figure 3).
-

Figure 3. Ligand substitution in tetrahedral Ni(CO)₄ proceeds through a dissociative mechanism, with CO dissociation as the rate-determining step, as evidenced by first-order kinetics (−d[Ni(CO)₄]/dt = *k*₁[Ni(CO)₄]) independent of incoming ligand. The transient Ni(CO)₃ intermediate rapidly reacts with incoming ligands (L = CO, PR₃) or undergoes isotopic exchange with *Ni(¹³CO)₄ in the gas phase. While recombination with CO faces minimal barriers, solvent coordination and steric effects of bulky ligands can significantly alter the reaction pathway energetics.
Substitution in Different Electron Configurations
1. 16-Electron Complexes (e.g., Pt(II), Pd(II), Ni(II))
- Undergo associative substitution due to coordinative unsaturation.
-
Factors influencing rates:
-
Nucleophile dependence: nucleophile dependence for soft, polarizable Pt(II) follows the trend PR₃ > py ≈ NH₃ > Cl⁻ > H₂O > OH⁻, spanning a rate range of approximately 10⁵.
-
Leaving group ability: decreases in the order NO₃⁻ > H₂O > Cl⁻ > Br⁻ > I⁻ > N₃⁻ > SCN⁻ > NO₂⁻ > CN⁻, reflecting the stability of the five-coordinate intermediate formed upon nucleophilic attack
-
Metal reactivity trend: rates decreasing in the order Ni(II) > Pd(II) ≫ Pt(II), a trend attributed to the varying ease of forming 18-electron five-coordinate intermediates
-
Trans Influence (Thermodynamic Effect)
-
Definition: The ability of a ligand to weaken the metal-ligand bond trans to itself in the ground state (static effect).
Cause: Strong σ-donation or π-backbonding from the ligand reduces electron density along the trans axis.
Trend:
R₃Si⁻ > H⁻ ≈ CH₃⁻ ≈ CN⁻ ≈ CO/olefins > PR₃ > NO₂⁻ ≈ I⁻ ≈ SCN⁻ > Br⁻ > Cl⁻ > NH₃ ≈ H₂O
Strong σ-donors (H⁻, CH₃⁻) and π-acceptors (CO, C₂H₄) have the greatest trans influence.
-
- Trans Effect (Kinetic Effect)
Definition: The ability of a ligand to accelerate substitution of the ligand trans to itself.
Cause:Ligands that are strong σ-donors and π-acceptors (CO, CN⁻, H⁻) show the strongest trans effect.
-
-
-
σ-donation: Weakens the trans M–L bond in the transition state.
-
π-acceptance: Stabilizes the 5-coordinate intermediate by delocalizing electron densityTrend:
CN⁻ ≈ CO ≈ C₂H₄ ≈ H⁻ > PR₃ > NO₂⁻ > I⁻ > Br⁻ > Cl⁻ > NH₃ > H₂OLigands that are strong σ-donors and π-acceptors (CO, CN⁻, H⁻) show the strongest trans effect.
Trend:
CN⁻ ≈ CO ≈ C₂H₄ ≈ H⁻ > PR₃ > NO₂⁻ > I⁻ > Br⁻ > Cl⁻ > NH₃ > H₂O
-
-
2. 17-Electron Complexes: Hyper-Reactive Radical Intermediates
- 17-electron complexes exhibit dramatically faster ligand substitution rates than their 18-electron counterparts due to their open-shell radical character.
-
Key Features:
-
-
Extreme Lability: Substitution occurs up to 10³–10¹⁰ times faster than in 18e⁻ complexes.
-
Example: V(CO)₆ substitutes CO with PPh₃ at –70°C in 90 min, while [V(CO)₆]⁻ (18e⁻) is inert even in molten PPh₃.

-
- Mechanism: Initially, a dissociative pathway (loss of CO to form a 15e⁻ intermediate) was assumed. However, molecular orbital theory explains why associative substitution dominates:
-
Incoming ligand forms a 19e⁻ intermediate.
-
Net stabilization: 2 electrons fill a bonding orbital, while only 1 enters an antibonding orbital.
-
Some stable 19e⁻ complexes exist (e.g., [CpFe(CO)₂]•) if the extra electron resides on a ligand.
-
- Experimental Evidence:
-
Steric effects: Bulky ligands (e.g., P(i-Pr)₃) slow substitution.
-
Hard bases (pyridine, THF) often cause disproportionation instead of substitution.
-
Persistent radicals like Mn(CO)₃(PR₃)₂ confirm associative pathways.
-
-
3. 18-Electron Complexes: Hyper-Reactive Radical Intermediates
- 18e⁻ complexes are coordinatively saturated and substitution requires breaking strong M–L bonds.
-
Challenges:
-
High Activation Energy: (25–37 kcal/mol, close to M–L bond energies).
- Extremely slow rates: CpRh(C₂H₄)₂ exchanges ethylene ~10¹⁴× slower than 16e⁻ Rh(acac)(C₂H₄)₂.
-
Activation Strategies
| Method | Example | Effect |
|---|---|---|
| Photolysis | Cr(CO)₆ + hν → Cr(CO)₅ + CO | Cleaves M–CO bonds |
| Lewis base catalysis | Fe(CO)₅ + R₃P=O → Fe(CO)₄(PPh₃) | Lowers ΔG‡ |
| Redox initiation | [Ni(CO)₄] → [Ni(CO)₃]⁺ + e⁻ | Generates 17e⁻ intermediate |
| Radical chains | HRe(CO)₅ + hν → Re(CO)₅• + H• | Accelerates via radicals |
- Condition Sensitivity:
-
HRe(CO)₅ + PBu₃:
-
With light: Substitution in minutes.
-
Dark, ultra-pure: No reaction after 2 months.
-
-
Fe(CO)₅:
-
Pure: Unreactive below 90°C.
-
Trace impurities: Reacts at 60°C.
-
-
Catalyzed Substitution Pathways
A. Electorn Transfer Catalysis
- Four mechanisms (SON1 , SOE1 , SRE2 , SRN1 ) via electrochemical/chemical redox.
B. Radical Chain Mechanisms
-
Observed in HRe(CO)₅ → HRe(CO)₃L₂:
-
Exclusive disubstitution (no mono-substituted product).
-
Oxygen inhibition (quenches radicals).
-
Photocatalyzed by Re(CO)₅•.
-
C. Other Assisted Pathways
| Strategy | Example |
|---|---|
| Brønsted/Lewis acids | Protonation of CO to labilize it |
| Nucleophiles (R₃NO, MeO⁻) | Attack on CO to form unstable adducts |
| Sonochemistry | Ultrasound-induced ligand dissociation |
Key Takeaways
-
16e⁻ complexes: Fast, associative, controlled by trans effects
-
17e⁻ complexes: Radical-driven, extremely labile
-
18e⁻ complexes: Require activation (light, redox, catalysts)
-
Design principles: Electron count and conditions dictate mechanism